MEDICINA INTERNA: VOLUME PRIMO
Titolo originale: Internal Medicine 2° edizione 1998. Editoriale
Grasso Bologna.
Capitolo
N° 75. Malattia polmonare
interstiziale diffusa
A cura
del DR: Ruy V. Lourenco, Christopher S. Garrard Curatore della edizione
Italiana delle malattie del polmone: Prof. Antonio Strano Professore ordinario
di medicina interna Università Tor Vergata di Roma
Le
malattie polmonari interstiziali sono caratterizzate da alterazioni che
coinvolgono inizialmente l'epitelio alveolare e le cellule endoteliali,
l'interstizio di supporto e, in misura minore e variabile, altre strutture
vascolari e vie aeree. Esse formano un eterogeneo gruppo di patologie, alcune
delle quali conseguenti a cause facilmente identificabili, mentre in altre la
causa sottostante resta oscura. Sebbene molto meno comuni delle malattie delle
vie aeree come la BPCO, le malattie polmonari interstiziali
costituiscono un importante gruppo di malattie associate a sintomi invalidanti
ed ad un rapido deterioramento clinico. I termini polmonite ed alveolite sono
usati di frequente per descrivere il processo flogistico che coinvolge le
cellule alveolari in molte malattie interstiziali. In alcune di queste malattie
un'anomala disposizione del collagene segue il processo flogistico acuto, in
maniera tale che il termine fibrosi polmonare è usato quasi in maniera
intercambiabile con quello di malattia polmonare interstiziale.
Classificazione eziologia. È conveniente e clinicamente corretto
dividere la fibrosi polmonare diffusa in due grandi categorie: quella ad
eziologia nota, che rappresenta circa un terzo dei casi, e quella ad eziologia
ignota. Man mano che le procedure diagnostiche diventano più sensibili e
specifiche, l'ultimo gruppo dovrebbe diventare meno comune. Nel gruppo da cause
conosciute, si possono ancora distinguere tre grandi sottogruppi di fibrosi polmonare
interstiziale:
1. quello determinato da granulomi estesi
2. quello determinato da essudati polmonari cronici
3. quello dovuto a polveri fibrogene inorganiche.
Diverse
entità patologiche contribuiscono ad ogni sottogruppo e sono elencate nella
tabella 75.1. Dopo l'esclusione delle forme da causa conosciuta,
rimane un vasto gruppo di malattie polmonari interstiziali fibrotiche (tab. 75.2). In questo gruppo vi sono malattie con
caratteristici aspetti istopatologici, che sono diventate note sotto gli
pseudonimi di: fibrosi polmonare idiopatica (FPI), fibrosi interstiziale
diffusa da causa ignota, e alveolite criptogenetica fibrotica (ACF). La
sindrome di Hamman-Rich, è un entità morbosa rarissima, e caratterizzata da
omogeneità temporale della patologia, con una forma fibrotica, rapidamente
progressiva di FPI. In questo capitolo si userà il primo di questi sinonimi,
fibrosi polmonare idiomatica (FPI) Dapprima saranno considerati alcuni
aspetti della fibrosi polmonare diffusa comuni a tutte le malattie fibrotiche,
senza tener conto della causa, e poi le singole malattie saranno discusse di
volta in volta in base a patogenesi, storia clinica, fisiopatologia, diagnosi,
e terapia.
|
Tabella 75.1
Malattie polmonari Interstiziali a causa conosciuta Granuloma
polmonare esteso Sarcoidosi (•) Berilliosi Alveolite
allergica estrinseca Essudati
polmonari cronici Edema
polmonare cardiogeno cronico Inalazione di
gas, fumi e vapori Reazioni ai
farmaci Radiazioni Uremia Polmoniti
virali, batteriche, fungine, parassitarie Inalazione di polvere
inorganica fibrogenica Amianto Silice Talco
|
Tabella 75.2
Malattie polmonari Interstiziali a causa sconosciuta Fibrosi polmonare idiopatica (alveolite
fibrosante criptogenetica) e varianti istologiche (PIC, PID, PIG, PIL) Malattie del collagene e autoimmuni Vasculiti sistemiche Granuloma eosinofìlo Malattia polmonare eosinofila Linfangioleiomatosi Disturbi ereditari PIC: Polmonite interstiziale comune, PID:
polmonite interstiziale desquamativa, PIG: polmonite interstiziale a cellule gicanti, PIL: polmonite interstiziale linfoide. Iinfoide |
Segni
radiografici
La
distribuzione radiografica della malattia interstiziale nel polmone è spesso
caratteristica della patologia di base La fibrosi polmonare nel lobo superiore
si osserva nella silicosi e nella spondilite anchilosante. Le reazioni
granulomatose osservate nella sarcoidosi, sono di solito distribuite
uniformemente in tutto il polmone, ma, quando si stabilisce una fibrosi
cronica, a nota spesso una predilezione per i lobi superiori Una fibrosi,
preferibilmente nel lobo inferiore si osserva nell’asbestosi, nella FPI, nella
fibrosi associata a sclerodermia, nell’artrite reumatoide e nel lupus
eritematoso sistemico e nelle reazioni a farmaci tossici. Questi ultimi esempi
in accordo con il fatto che la perfusione sanguigna è maggiore nelle regioni
inferiori del polmone, sono prove di un fattore eziologico di origine ematica,
sia esso un immunocomplesso circolante od un farmaco tossico. La fibrosi da
asbestosi e la FPI si riconoscono perché risparmiano le regioni polmonari
centrali, sicché si realizza un coinvolgimento periferico e subpleurico molto
più vasto. Radiograficamente, la malattia polmonare interstiziale può assumere
diverse forme, manifestandosi variamente come opacità reticolari, nodulari o
reticolonodulari, dense masse fibrotiche o cicatrici lineari. Un quadro
reticolare compare come fini ombre lineari distribuite irregolarmente spesso
formanti anelli sottili che racchiudono spazi aerei. Un quadro nodulare
consiste di opacità multiple, di solito rotonde di varie dimensioni, da meno di
1 mm a più di 10 mm Un quadro
reticolo-nodulare comprende entrambe le componenti. Alcune malattie
interstiziali producono un aspetto a superficie vetrosa in cui i campi
polmonari sono di radiodensità aumentata con fini granulosità. Tardivamente, in
certe malattie polmonari interstiziali, i campi polmonari assumono un aspetto
ad alveare, in cui sono osservabili molte ombre ad anello tra 5 e 8 cm di diametro, con
pareti sottili da 0,5 ad 1 mm. Alcuni autori descrivono le
modificazioni radiografiche in termini di quadri interstiziali ed aerei. Questa
descrizione presume una correlazione ideale tra l'aspetto radiografico e le
variazioni istologiche. La descrizione delle ombre reticolari o nodulari,
comunque, non è basata su questo assunto. La fibrosi del lobo superiore, con
perdita di volume polmonare può innalzare la piccola scissura orizzontale e se
si verifica una contrattura unilaterale, causa la deviazione della trachea. La
fibrosi del lobo inferiore può essere accompagnata da una progressiva
elevazione del diaframma e da una perdita di volume polmonare. In qualsiasi
regione del polmone si possono sviluppare bolle enfisematose, che sono
comunemente segno di silicosi o di sarcoidosi avanzata. La maggior parte dei
pazienti presenta brevità del respiro durante gli esercizi fisici che, con la
progressione della malattia, si palesa perfino a riposo. Il grado di dispnea
può essere estremo e non proporzionato alla gravità delle modificazioni
radiologiche. L'opposto può essere osservato nella sarcoidosi, in cui,
nonostante gli impressionanti cambiamenti radiografici dei polmoni, la dispnea
può essere minima. Una tosse secca e non produttiva è un sintomo frequentemente
associato e spesso d'esordio. Sintomi sistemici aspecifici, come affaticamento
e la perdita di peso, possono essere osservati in circa la metà di casi di
malattie polmonari interstiziali. La febbre può essere presente, specialmente
nei casi acuti e rapidamente progressivi di FPI, proteinosi polmonare alveolare
e nelle malattie del collagene. I sintomi respiratori dovuti a una compromissione
interstiziale dei polmoni possono essere caratteri d'esordio di malattie del
collagene come artrite reumatoide o la sclerodermia. In queste malattie comunque, i sintomi correlati ad altri
sistemi, come artrite, rash cutanei o anemia, sono di solito dominanti e
precedono i sintomi respiratori.Una globale ed attenta anamnesi sociale,
lavorativa e farmacologica è essenziale, se si deve riconoscere una malattia
polmonare interstiziale assodata all'esposizione a polveri organiche o non e a
certi farmaci. L'esame obbiettivo rivela spesso una respirazione rapida e poco
profonda, con il reclutamento di muscoli accessori negli stadi più tardivi
della fibrosi polmonare. L'ippocratismo digitale, che in alcuni casi può
precedere lo sviluppo dei sintomi respiratori, suggerisce fortemente FPI,
asbestosi o la coesistenza di malattie quali il carcinoma broncogeno o le
bronchiectasie. L'ascoltazione del torace nei pazienti con FPI rivela
tipicamente fini crepitazioni predominanti alle basi. In alcune malattie polmonari
interstiziali, i reperti auscultatori polmonari possono essere poco importanti
od apparire solo tardivamente sotto forma di sibili o ronchi indicanti un
interessamento delle vie aeree. Lo sviluppo di un cuore polmonare di solito è
una manifestazione di malattia polmonare in fase terminale ma può essere
presente in fasi più precoci della sclerodermia o nelle malattie del collagene,
in cui l'interessamento dei vasi polmonari è un importante aspetto del processo
patologico.
Patogenesi
Senza
una particolare specificità per l'agente patogeno, la risposta del polmone al danno segue un disegno ben riconoscibile
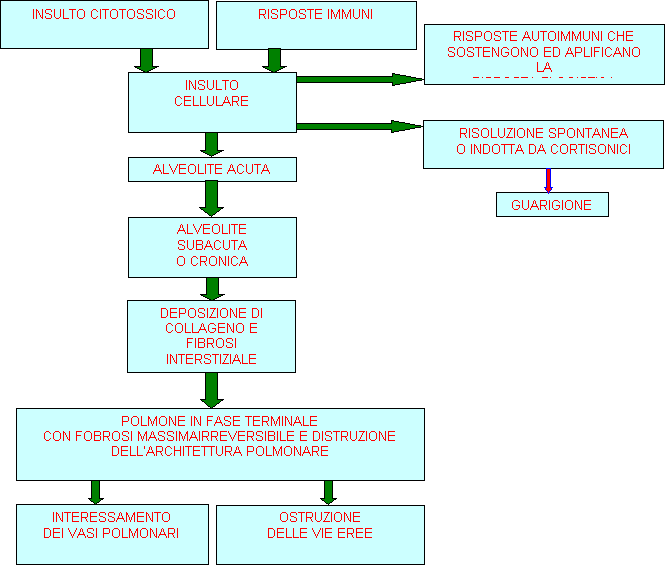
Si possono presentare diversi stadi nello sviluppo della fibrosi polmonare a
cominciare da una alveolite acuta caratterizzata da un aumento del numero dei
macrofagi alveolari e dei linfociti nell'interstizio polmonare (fig 75.1) In base alla cronicità dell'infiammazione
interessante il polmone o all'efficienza dei meccanismi di difesa del polmone,
l'alveolite acuta può progredire in una forma cronica di alveolite
caratterizzata da un danno al pneumocita di I tipo con proliferazione di pneumociti
di II tipo. La continuità degli strati della membrana
basale tra il pneumocita e la cellula capillare endoteliale, sembra essere
essenziale per il mantenimento della normale architettura alveolare durante un
successivo processo di guarigione. Quando il processo infiammatorio continua,
si verifica fibrosi polmonare con quantità, tipi e distribuzione di fibre
collagene anormali, fibre che sono prodotte dai fibroblasti attivati. Lo stadio terminale del polmone
è caratterizzato dalla completa perdita della normale struttura alveolare. In
tutto il polmone malato appaiono degli spazi cistici non funzionanti. Questo
stadio, è il punto di arrivo comune a molti processi infiammatori
interstiziali.
Figura 75.1 Decorso e sviluppo della fibrosi
polmonare.
|
INSUFFICIENZA RESPIRATORIA IPPOSSIMIA E
CUORE POLMONARE |
Fisiopatologia della fibrosi polmonare
E
tipico un restringimento di tutti i volumi polmonari con la riduzione della capacità vitale (CV),
della capacità funzionale residua (CFR) e della capacità polmonare
totale (CPT), sebbene il volume residuo (VR) possa essere meglio
conservato. Ripetute misurazioni dei volumi polmonari e della diffusione di CO
(DLCO) possono essere usate per controllare la progressione della
malattia. Sono stati proposti per l’identificazione dei disturbi iniziali test
più sensibili, come, per esempio, la differenza dell'ossigeno alveolo-arterioso
[P(A-a)02:] durante l'esercizio, ma, probabilmente, essi
offrono scarsi vantaggi rispetto alle semplici misurazioni spirometriche dei
volumi in una malattia già stabilita. E chiaro che si può osservare ostruzione
delle vie respiratorie nella fibrosi polmonare interstiziale. Per esempio,
nella sarcoidosi in fase avanzata, nel 25% dei pazienti si può avere un
rapporto tra volume espiratorio forzato a 1 sec e capacità vitale forzata (FEV
1,o/CVF%) ridotto. I test della funzione delle piccole vie aeree (tasso di
flusso espiratorio a bassi volumi polmonari e la pendenza del plateau della
concentrazione dell'azoto espirato durante la III fase del washout dell'azoto di un singolo
respiro), possono essere anomali negli stadi iniziali di molte malattie
polmonari fibrotiche, compresa l'asbestosi, la pneumoconiosi del minatore e la
polmonite da ipersensiblità. Di solito, col progredire della riduzione dei
volumi polmonari segue un danneggiamento della capacità di diffusione del
monossido di carbonio (DLCO). Inizialmente si pensava che questo fosse
solo un fenomeno di blocco alveolo-capillare; comunque, i disturbi della
distribuzione ventilatoria dovuti a coinvolgimento delle vie aeree
probabilmente costituiscono il difetto più osservato. Analogamente,
l'ipossiemia arteriosa è il risultato di un cattivo rapporto
ventilazione/perfusione con una componente minore di blocco alveolo-capillare.
Un aumento dell'elasticità ed una diminuzione della compliance dei polmoni sono
segni tipici della fibrosi polmonare. Una parte dell'aumento dell'elasticità,
tuttavia, è attribuibile ad una diminuzione del numero degli alveoli ed alla
conseguente perdita di volume polmonare. I gas arteriosi di solito mostrano
riduzioni della PaO2, con associate riduzioni della PaCO2; un
PH normale o una lieve alcalosi respiratoria e un aumento della P(A-a)02.
Sebbene un'ipossiemia severa possa essere osservata negli stadi più tardivi di
malattia polmonare interstiziale, può essere anche presente nelle fasi iniziali
di alcune di esse, come la proteinosi alveolare polmonare, la sindrome di
Goodpasture e l'esposizione acuta a fumi tossici. I pazienti con fibrosi
polmonare hanno molti atti ventilatori al minuto, a riposo e durante esercizio,
che danno luogo ad un anormale abbassamento della PaCO2; arteriosa.
Questa iperventilazione si verifica malgrado un aumento del lavoro meccanico
necessario per la minore elasticità del polmone fibrotico. E stato suggerito
che il più alto livello di ventilazione sia dovuto ad un aumento degli impulsi
afferenti che partono dal polmone o dai muscoli respiratori. I recettori di
stiramento od i recettori J possono invilupparsi nel tessuto fibroso oppure un
inappropriato rapporto lunghezza/tensione con le giunture dei muscoli può
svilupparsi a causa della ridotta compilarne polmonare. L'aumento dell'attività
afferente trasmessa al cervello stimola i centri respiratori e aumenta
l'attività del sistema nervoso efferente. Questo aumento causa
un'iperventilazione che può contribuire alla sensazione di dispnea che è
avvertita da questi pazienti.
Aspetti immunologici
L'associazione fra fibrosi
polmonare, disturbi autoimmuni e malattie collagene-vascolari ha suggerito che
anticorpi circolanti non specifici di un organo possano essere coinvolti come
agente causale. Il fattore reumatoide può
essere presente in percentuali significative in malattie quali la pneumoconiosi
del minatore (dal 30 al 40% dei casi. Con fibrosi massiva progressiva) e
la fibrosi polmonare idiopatica (15% dei casi) e ha posto degli interrogativi
riguardo all'esatto rapporto esistente fra meccanismi immunologici e fibrosi
polmonare. Autoanticorpi, in particolare il fattore antinucleare, sono stati
dimostrati in percentuali aumentate in associazione a fibrosi polmonare
idiopatica (40% dei casi), asbestosi (25% dei casi),
pneumoconiosi del minatore con fibrosi massiva (74% dei casi) e silicosi (40% dei casi).
Sebbene le percentuali del fattore antinucleare siano circa un decimo di quelle
trovate nelle malattie del collagene, esse sono significativamente più alte di
quelle trovate in popolazioni normali. Queste scoperte ed altri dati
sperimentali suggeriscono che gli anticorpi non organo-specifici possano sia
avere un ruolo causale nelle malattie riconosciute come autoimmuni che accelerare
la progressione della fibrosi nelle altre malattie interstiziali.
Malattie polmonari interstiziali a causa nota
Grandi granulomi. Sarcoidosi: la sarcoidosi è caratterizzata dalla presenza di granulomi a
cellule epiteliali non caseosi in molti sistemi d'organo, dei quali uno dei più
frequentemente interessati è il polmone. Ulteriori dettagli sugli aspetti
polmonari ed extrapolmonari della sarcoidosi si trovano nel capitolo 77.
Berilliosi. L'inalazione di
berillio come metallo o come sale può causare una polmonite chimica acuta (berilliosi
acuta) o una reazione granulomatosa cronica di tipo sarcoide (berilliosi
cronica).La maggior parte dei casi registrati comparve tra il 1943 ed n
1955 quando il berillio veniva usato estesamente nella manifattura di luci
fluorescenti. Oggi la maggior parte dei casi è collegata all'uso di berillio in
aviazione e nelle industrie ad energia nucleare. I dati disponibili indicano
che una reazione d'ipersensibilità è responsabile del processo infiammatorio
attivo. La berilliosi acuta è caratterizzata da un grave edema polmonare. Gli
alveoli sono pieni di coagulo fibrinoso e di un essudato ricco di
polimorfonucleati. Le pareti alveolari sono piene di cerule plasmatiche, linfociti
e depositi di proteine.Il processo di guarigione, annunziato dalla
proliferazione dei macrofagi alveolari, di solito progredisce con gradi diversi
di fibrosi interstiziale. La
berilliosi cronica è caratterizzata dallo sviluppo di grandi granulomi polmonari
che sono indistinguibili da quelli della sarcoidosi. Oltre ad una terapia di
supporto con O2, dovrebbero essere somministrati cortisonici non appena è
diagnosticata una berilliosi, sebbene una piccola proporzione di casi possa non
rispondere, sviluppando una fibrosi progressiva. La terapia può dover essere
continuata a lungo, come per la sarcoidosi. Le misure di prevenzione rimangono
gli aspetti più importanti nel controllo di questa malattia.
Alveolite allergica estrinseca: in alcuni individui, l'inalazione di
polveri organiche provenienti dall'ambiente familiare o di lavoro da luogo a
reazioni di ipersensibilità negli alveoli. In contrasto con la risposta
allergica delle vie aeree asmatica mediata da una reazione immune immediata di
tipo I o da anticorpi non precipitanti, l'alveolite è mediata da una
reazione di anticorpi precipitanti (tipo III). Le due reazioni, tuttavia, coesistono
nello stesso individuo. Come la sarcoidosi e la berilliosi, questo gruppo di
alveoliti allergiche, provoca reazioni granulomatose nel polmone.Una
trattazione completa di questo argomento è contenuta nel capitolo 78.
Essudati polmonari cronici: Edema polmonare cronico. L’accumulo di liquido nell’interstizio
polmonare, negli spazi perivascolari, nei linfatici ed infine negli spazi
alveolari può svilupparsi dopo aumenti della pressione capillare intracapillare
(edema polmonare cardiogenico) o dopo un cambiamento della permeabilità
capillare polmonare (edema polmonare non cardiogenico). L'edema polmonare
cardiogenico è una ben nota complicanza dell'insufficienza ventricolare,
sinistra o della malattia della valvola mitrale. L'edema polmonare non
cardiogenico può essere visto come il risultato di una vasta gamma di danni
tossici al polmone, compresi gas e vapori inalati, farmaci, tossine batteriche e radiazioni. L'esposizione cronica a questi
agenti può alla fine condurre alla fibrosi polmonare. L'edema polmonare è
discusso nel capitolo 70.
Gas, fumi e vapori inalati: Esempio di agenti inalati noti per
produrre malattie polmonari interstiziali sono elencati
nella tabella 75.3.
Ossigeno: l’effetto tossico di alti livelli di
ossigeno sui polmoni di neonati e adulti è ben riconosciuto. La maggiore
conoscenza della tossicità da O2, ha portato a una maggiore
attenzione circa il controllo della soniministrazione di O2,
Sebbene la maggior parte delle osservazioni di tossicità sia stata correlata
all'uso di O2, al 100%, è probabile che perfino livelli
moderati di O2, (50-(6O%) siano tossici. L'esperienza clinica
indica che l’-02, a concentrazioni del 40% o meno è raramente
tossico. Danni" ai pneumociti di I tipo e proliferazione di pnumociti di II tipo sono le
manifestazioni iniziali di tossicità Lo sviluppo di un'emorragia intralveolare,
la formazione di' una membrana ialina e la proliferazione fibroblastica negli spazi
aerei alveolari e interstizio portano a fibrosi polmonare. La proliferazione
dei capillari alveolari può essere considerata un equivalente della
proliferazione dei capillari retinici osservata nella fibroplasia
retrolenticolare del neonato I caratteri patologici della tossicità da O2, sono molto simili a quelli osservati
nella polmonite da raggi, particolarmente per quanto riguarda effètti sulle
cellule endoteliali. Non è chiaro il meccanismo che determina la tossicità
dell'ossigeno, ma si è visto che l'ipossiemia interferisce con la produzione di
fosfato ad alta energia (ADP, ATP) È stato suggerito che una riduzione dei
processi metabolici dei pneumociti di tipo II, ad esempio, possa
compromettere la produzione del surfactante e le funzioni riparative di queste
cellule. La fagocitosi da parte di macrofagi alveolari può essere pure
compromessa. L'interesse si è localizzato sul ruolo dei radicali liberi
superossido, che sono generati in presenza di iperosia e sono in grado di
distruggere l'integrità delle membrane cellulari. Studi su animali hanno
dimostrato una tolleranza da adattamento all'iperossia in risposta ad
incrementi graduali della concentrazione di O2, inalato. Tale
adattamento è probabilmente dovuto a un'aumentata disponibilità di superossido dismutasi,
che riduce la tossicità dei radicali superossido. Interazioni sinergiche sono
state osservate tra la tossicità da O2; ed altri agenti lesivi come
l'erbicida paraquat, radiazioni e bleomicina. Una maggiore attenzione ai rischi
potenziali di tossicità dell’O2, ha portato a un approccio più
critico alla prescrizione dell’ossigenoterapia. Sia nella sindrome da membrane
ialine dei prematuri che nell'ARDS, l'utilizzazione di un tubo endotracheale e
l'applicazione di una pressione positiva continua nelle vie aeree ha permesso,
correggendo lo squilibrio V/Q, di impiegare concentrazioni di.O2,
inspirato molto inferiore. I cortisonici sono stati prescritti nei casi di
sospetta tossicità da O2, sebbene i benefici di un tale trattamento
non siano comprovati.Gas come il cloro e l'anidride solforosa sono estremamente
irritanti e causano un edema polmonare acuto, se presenti nell’atmosfera a
concentrazioni abbastanza alte. Raramente
l'esposizione a lungo termine a cloro od anidride solforosa può causare
una fibrosi cronica. Una prolungata esposizione a bassi livelli di CI ed SO2, può anche indurre una
disfunzione polmonare che può essere
evidenziata con i test di funzionalità polmonare.
Altri vapori: Vapori di diisocianuro di toluene prodotti durante la manifattura
di poliuretani possono indurre una
bronchite ed un asma acuti. Con esposizioni croniche a bassi livelli si
possono sviluppare una polmonite interstiziale e perfino una fibrosi polmonare.
Aerosol di sonato di rame usati m viticoltura per sopprimere le infezioni
(fungine, provocano una reazione granulomatosa interstiziale probabilmente su
base immunologica.
Tabella 75.3 esempi di agenti inalati
che causano malattie polmonari interstiziali
|
||
|
Stato
fisico Gas: Fumi: Metallici: Vapore: Aspirazione: Aereosol |
Agente Ossigeno. Cloruri. Biossido di zolfo. Ossidi di
zinco. Manganese,
carminio. Ferro e
nichel. Mercurio. Resine
termoindurenti. Diisocianato
di toluetene. Lipidi, Acidi
gastrici. Piretro |
Fonte Somministrazione Terapeutica. Incidenti
industriali. Sulfurei. Saldatura,
fusione e Galvanizazione. Esposizione
accidentale. Industria
della gomma. Poliuretano e
gomma sintetica. Cultura e
gocce nasali. Insetticidi |
Grassi. Oli di origine minerale, vegetale o animale possono essere inalati,
inducendo una malattia polmonare interstiziale spesso detta polmonite lipoide
esogena (V.cap. 73).
Aspirazione dei succhi gastrici (sindrome di mendelson) L’aspirazione nei polmoni di acidi gastrici provoca un edema
polmonare acuto con conseguenti ipossiemia acuta e ARDS (V. cap. 73)
Fumi metallici. L'inalazione
di vapori di mercurio provoca disturbi acuti sistemici, come dolore addominale,
diarrea e sintomi a carico del SNC. La tossicità respiratoria si
estrinseca in severe tracheiti, bronchiti e polmoniti.gli ossidi di manganese generati durante la manifattura
delle batterie a secco producono pure un'irritazione del tratto respiratorio
superiore e polmonite che, con l'esposizione cronica può portare a una fibrosi
polmonare.
Reazioni
Indotte da farmaci. Reazioni avverse da
farmaci che provocano una significativa morbidità e mortalità sono presenti in circa il 10% dei pazienti ospedalizzati sotto terapia.Una
parte di queste reazioni avverse interessa i polmoni colpendo sia le vie aeree
che il parenchima. Numerosi farmaci, in particolare antibiotici come penicillina, tetraciclina
eritromicina e cefaloridina, sono assodati ad una risposta asmatica e non
saranno ulteriormente considerati in questo contesto. L’interessamento del
parenchima polmonare può assumere forme diverse, incluse alveoliti acute o
croniche polmoniti eosinofile e da ipersensibilità e reazioni granulomatose. Le malattie interstiziali indotte da farmaci si presentano molto spesso
in maniera insidiosa, con il paziente che si lamenta di tosse e dispnea. Questi
sintomi si possono sviluppare rapidamente, se vi è una reazione infiammatoria acuta nel polmone. La radiografia del torace di solito
rivela infiltrati interstiziali ed i test fisiologici di funzionalità polmonare
dimostrano alcuni gradi di restrizione polmonari e difetti associati nello
scambio e nella diffusione dei gas.Queste
modificazioni non sono specifiche ed ulteriori indagini, possibilmente
culminanti con una biopsia polmonare possono essere richieste per la
formulazione della diagnosi. Perfino quando si sospetta una pneumopatia indotta
da farmaci, è spesso difficile stabilire una relazione causale, perché la
malattia sottostante per la quale il farmaco è stato prescritto può causare
essa stessa una malattia interstiziale, come nel caso degli agenti citotossici usati nel trattamento dei disordini
linforeticolari. Non si
dovrebbe comunque risottoporre il paziente al trattamento con il particolare
farmaco sospettato per confermare la diagnosi di pneumopatia indotta da
farmaci. Un elenco di farmaci
che causano malattie polmonari interstiziali è riportato nella tabella 75.4. Numerose categorie di farmaci sono
frequentemente implicate nella genesi di malattie polmonari interstiziali e i
più importanti saranno esaminati individualmente.
Agenti
citotossici ed immunosoppressori. Il gruppo dei
farmaci citotossici immunosoppressori è fra quelli più frequentemente implicati
nelle malattie polmonari indotte da farmaci.
Bleomicina. La bleomicina può
indurre una fibrosi polmonare nel 5-10% dei pazienti che la ricevono, sebbene
alcune statistiche abbiano riportato un'incidenza quasi fino al 50%.
L’incidenza delle malattie interstiziali è dose dipendente ed è rara con una
dose totale inferiore ai 50 mg. La via di somministrazione sembra avere poca o
nessuna importanza sulla comparsa della pneumopatia, sebbene vi possa essere
qualche indicazione che l’infusione continua di bleomicina sia meno tossica dei
boli endovenosi. La tossicità può essere aumentata da precedenti trattamenti
con bleomicina, da un’insufficienza renale indotta dalla stessa (che viene
eliminata per via renale) e dalla combinazione con altri agenti citotossici, da
una radioterapia applicata ai campi polmonari e da una concomitante
somministrazione di O2. I caratteri istologici della
fibrosi indotta da bleomicina e da busulfano sono virtualmente indistinguibili.
Emorragie ed essudati fibrinosi con occasionale ialinizzazione sono osservati
entro lo spazio aereo alveolare. Gli pneumociti di tipo I contengono bizzarri
nuclei ipercromatinici e l’interstizio è infiltrato da linfociti,
plasmacellule, eosinofili e fibroplasti; si possono riconoscere aree di fibrosi
dense ed organizzate.
|
Tabella 75.4 Farmaci che possono essere causa di malattie polmonari interstiziali |
|
Agenti citotossici e immunosoppressori. Bleomicina Busulfano
|
Antimicrobici Eritromicina Etionamide Griseofulvina Isoniazide Nitrofurantoina Acido paraminosalicilico Penicillina Sulfamidici salicilazosulfopiridina |
Agenti endocrinologici Cortisoni Clorpropamide Estrogeni Estratti pituitarici |
Miscelania Amiodarone Amitriptilina Beclometasone dipropionato Clordiazepossido Cromolin sodico Sali d’oro Impramina Mezzi di contrasto contenenti iodio Marijuana Medisergide Olio minerale Chinidina Penicillamina Alcune fenotiazine Pindololo Ioduro di K+ Practololo Warfarin |
|
Analgesici non narcotici Acido acetilsalicilico Ibuprofene fenilbutazone |
Analgesici narcotici Eroina Metadone Proposifene |
Antidepressivi Idroclorotiazide Esametonio Mecamilamina Pentolinio |
Anticonvulsivanti Carbamazepina Clonazepam Fentoina |
I test fisiologici di
funzionalità polmonare forniscono un utile metodo per scoprire presto una
tossicità;: polmonare e prima comparsa
dei sintomi o di una radiografia del torace anomala. Una progressiva riduzione
della capacità vitale, della capacità polmonare totale della diffusione
polmonare da monossido di carbonio (DLCO) possono comparire col
progredire della fibrosi. Può essere presente un’ipossiemia arteriosa.
I sintomi
conseguenti a una pneumopatia indotta bleomicina possono svilupparsi subito
dopo la somministrazione del farmaco o parecchi mesi dopo l'inizio del
trattamento. E comune una mancanza di fiato durante gli sforzi con una tosse
secca e improduttiva, sebbene alcuni pazienti possano esordire con febbre.
L'esame obbiettivo svela tachipnea e crepitazioni ad ambo le basi.
La radiografia del torace
rivela inizialmente un quadro reticolare diffuso coinvolgente i segmenti
basali; questo quadro evolve in un consolidamento alveolare più denso in tutti
campi polmonari. Se si stabilisce una fibrosi polmonare, l'infiltrato può
prendere un aspetto più nodulare. La scintigrafia polmonare al Gallio-67
può essere utile per dimostrare un processo flogistico acuto, ma non è
specifica e non può differenziare una tossicità polmonare da altri processi
flogistici.
La sospensione del trattamento con bleomicina può arrestare la progressione della tossicità
polmonare o può perfino essere seguita da un certo grado di risoluzione. In
alcuni casi vi può comunque essere un continuo deterioramento che alla fine
conduce allo scompenso respiratorio. I cortisonici possono combattere o
sopprimere le prime fasi del processo flogistico nel polmone da bleomicina, ma
è improbabile che migliorino una fibrosi stabilizzata.
Busulfan. Segni di
tossicità polmonare si manifestano in genere con un ritardo di 3-4 anni dopo
l'inizio della terapia con busulfan. L'incidenza di tossicità polmonare è
riportata dal 2% all'11% con caratteristiche simili a quella prodotta da altri
agenti citotossici. È stato riportato un effetto sinergico con le irradiazioni
polmonari.
Ciclofosfamide. L'incidenza di tossicità polmonare
indotta da ciclofosfamide è molto superiore rispetto a quella osservata con
altri citotossici. I caratteri di una malattia polmonare interstiziale si
possono sviluppare durante la terapia, ma appaiono più comunemente un anno dopo
il suo inizio. Nonostante la comparsa
di una fibrosi precoce, i cortisonici appaiono avere benefici effetti.
Metotressato.Il
metotressato, un antagonista dei folati, può
produrre una tossicità polmonare nel 10% dei pazienti trattati sebbene
alcune statistiche prospettiche non abbiano riportato casi di tossicità
polmonare. Lo sviluppo di tossicità
polmonare non è correlato alla dose somministrata e può risolversi
nonostante la continuazione del farmaco. Un'associata eosinofilia può riscontrarsi
in circa metà dei casi di tossicità polmonare. Linfoadenopatia ilare ed
affezioni pleuriche possono accompagnare i cambiamenti interstiziali.
Azatioprina. Esistono molti casi segnalati di polmonite da azatioprina.
Lincidenza di una polmonite interstiziale da azatioprina è ormai nota,
considerato che questo farmaco è usato in numerose condizioni neoplastiche e
non neoplastiche, inclusi malattie diffuse del connettivo e disordini
ematologiche non neoplastici. Tutta via la possibilità di una polmonite da
azatioprina in ogni paziente trattato con questo farmaco deve essere tenuta
presente. Essa viene metabolizzata in 6 mercaptopurina, per la quale ci sono
molti segnalazioni di polmonite interstiziale della varietà citotossica.
Tuttavia, la maggior parte di questi pazienti hanno ricevuto altri farmaci che
potenzialmente possono essere implicati nella polmonite.
Mitomicina.Circa
il 5% dei pazienti che vengono trattati con mitomicina può sviluppare una
tossicità polmonare entro 1-10 mesi dall'inizio della terapia. I caratteri
chimici e fisiopatologici sono specifici e gli effetti sono simili a quelli
prodotti da altri agenti citotossici. I cortisonici possono arrecare benefici.
Nitrosurea. La maggior
parte delle riportate tossicità polmonari sono state correlate all'uso di 2-3b
(2 cloroetil) –1 nitrosurea (BNCU) ma la clororotocina e il-metil-CCNU
(cloroetil citroetil nitrosureacùro) sono stati pure indiziati di provocare una
malattia polmonare interstiziale. La tossicità polmonare è dose dipendente, con
un'incidenza superiore al 50% nei pazienti che hanno ricevuto una
terapia intensa con alte dosi di BNCU. Segni e sintomi di tossicità polmonare
possono comparire da un mese a 5 anni dall'inizio della terapia, con un tempo
medio di esordio di circa un anno. Sono stati dimostrati effetti sinergia della
radioterapia sui campi polmonari e della contemporanea somministrazione di
altri agenti citotossici. I caratteri clinici sono simili a quelli osservati
con gli altri agenti citotossici, ad eccezione del fatto che la radiografia del
torace può con maggiore probabilità essere normale nelle fasi più precoci di
malattia. Quando i cambiamenti radiologici sono presenti, essi compaiono sotto
forma di infiltrati reticolari specialmente alle basi. Scansioni con Gallio-67
possono indicare una risposta flogistica attiva in assenza di modificazioni
radiografiche. I cortisonici sembrano avere poco effetto nel ridurre la
tossicità polmonare.
Antimicrobici. Penicillina,
acido para-aminosalicilico, sulfamidici, nitrofurantoina (menzionata nella
prossima sezione) ed isoniazide possono indurre infiltrati periferia
interstiziali, a volte associati a versamenti pleurici. I pazienti si
presentano con febbre, tosse, dispnea ed eosinofilia assoluta. Il processo
patologico sottostante non è chiaro, ma sono state riportate vasculiti
sistemiche in seguito a somministrazione di sulfamidici e penicillina,
indicando la possibile responsabilità di un processo immune. Reazioni asmatiche
con sibili e mancanza di respiro si verificano pure con numerosi antibiotici,
tra cui penicillina, tetracicline, eritromicina, neomicina, streptomicina,
cefatoridina ed etionamide. Nelle reazioni asmatiche la radiografia del torace
può mostrare solo una relativa iperlucentezza e iperinflazione.
Nitrofurantoina.
La più comune manifestazione di tossicità polmonare provocata da questo farmaco
è una polmonite acuta, che compare dopo alcune ore o giorni dall'inizio della
terapia con nitrofurantoina. Mentre la maggior parte delle flogosi si risolve
entro 1-4 giorni dalla fine della terapia, circa il 3% dei casi può progredire
verso una forma cronica. La forma cronica è associata a fibrosi polmonare, che
può avere caratteri istopatologici indistinguibili dalla FPI. La forma acuta si
presenta con febbre, raffreddore, dispnea e tosse. Ai Rx la forma acuta si
presenta con infiltrati sia interstiziali che aerei, insieme a versamenti
pleurici, mentre la forma cronica mostra dei cambiamenti interstiziali simili a
quelli osservati nella FPI. Anticorpi antinucleo (AAN) e livelli sierici
delle IgG possono essere aumentati, mentre i test di funzionalità polmonare
rivelano un quadro restrittivo con un'associata riduzione della DLCO. La risposta ai cortisonici di una
tossicità polmonare cronica può variare, ma può esservi un sorprendente
miglioramento nei quadri con un quadro istologico desquamativo.
Analgesi non narcotici.
Una stimolazione respiratoria centrale e un edema polmonare non cardiogenico
possono essere provocati da
un'overdose di acido salicilico, quando i livelli sierici di salicilato superano i 40 mg/dl. Si ritiene che i
livelli tossiti di salicilato aumentino la permeabilità delle membrane
endoteliali nei polmoni. Ai Rx si notano di solito infiltrati polmonari diffusi
in assenza di cardiomegalia. Questi infiltrati di solito si risolvono in alcuni
giorni con la normalizzazione dei livelli di salicilato. L'edema polmonare non
cardiogenico può essere tanto grave da provocare un'ARDS che richiede una
intubazione endotracheale e un supporto di pressione positiva nelle vie aeree.
Gli sforzi per aumentare la clearance del salicilato con una diuresi alcalina
forzata dovrebbero essere attentamente monitorati, per evitare un sovraccarico
idrico che può ulteriormente contribuire all'edema polmone. È stato riportato
che l'ibuprofene provoca una dispnea acuta associata ad opacità polmonari a
chiazze, e versamenti pleurici nei pazienti con malattie connettivali miste. In
seguito alla somministrazione di finilbutazone sono stati riportati edema polmonare e vasculiti
polmonari.
Analgesici narcotici. L'edema
polmonare è una complicazione comune dell'abuso di eroina endovenosa, che
avviene in circa 1/3 delle overdosi. L'edema polmonare si può
verificare anche con altri narcotici, incluso il metadone preso oralmente o per via nasale. Il suo preciso meccanismo
non è chiaro, ma è stato proposto un aumento della permeabilità endoteliale
secondario a deposizione di immunocomplessi. Altri meccanismi suggeriti
comprendono reazioni allergiche, un drenaggio linfatico compromesso e uno scompenso
del ventricolo sinistro. La radiografia del torace mostra diffusi infiltrati
indistinti di tipo sia interstiziale
che aereo che si risolvono di solito entro 24-48 ore. Emboli polmonari settici
possono portare alla formazione di ascessi polmonari multipli, in cui i
microrganismi consueti sono lo Suphylococcus
aureus e la Pseudomonas aeruginosa. In
alcuni pazienti, dopo ripetuti episodi di edema polmonare indotto da narcotici,
sono state riportate ronchiectasie che si manifestano clinicamente con tosse
persistente e produzione di un abbondante espettorato purulento. I contaminanti
dei narcotici iniettati endovena, come il talco, possono indurre reazioni
granulomatose che appaiono radiologicamente come infiltrati nodulari.
Anticonvulsivanti.
La fentoina e la carbamazepina sono state associate a occasionale comparsa di
febbre, tosse, dispnea, eosinofilia ed infiltrati polmonari. Questi sintomi si
risolvono dopo la sospensione dell'agente causale o in risposta a una terapia
cortisonica. Anche una linfoadenopatia ilare è stata descritta sia da sola che in associazione a processi
interstiziali.
Vari. Penicilamina.
La penicillamina può indurre una bronchiolite cronica obliterante in pazienti
trattati per malattie connettivali, come l'artrite reumatoide. La dispnea si
può sviluppare rapidamente e i test di funzionalità polmonare rivelano un
difetto ostruttivo irreversibile. La radiografia del torace può essere normale nel momento della comparsa dei
sintomi, ma più avanti possono comparire infiltrati interstiziali diffusi. Una
reazione da immunocomplessi è stata sospettata in alcuni pazienti con tossicità
polmonare indotta da penicillamina, portando all'uso benefico di immunosoppressori
e plasmaferesi. In parecchi casi la reazione da immunocomplessi indotta dalla
penicillamina ha indotto una sindrome simile a quella di Goodpastur. L'istotipo HLA-DRW2 è stato
osservato in gran parte dei pazienti con reazioni da immunocomplessi indotte da
penicillamina suggerendo una predisposizione ereditaria.
Sali aurei. I sali d'oro,
come l'aurotiomalato di Na, sono noti per produrre una polmonite e una fibrosi
polmonare in pazienti con artrite reumatoide. I dosaggi di sali aurei che
producono tossicità polmonare hanno un intervallo da 180 a1000 mg. I caratteri
di presentazione della tossicità aurea
includono dispnea, tosse, rash cutaneo, febbre, proteinuria ed
eosinofilia. La radiografia del torace mostra sia un quadro interstiziale che
uno misto, interstiziale ed aereo.Un meccanismo immune è stato sospettato,
portando all'uso di cortisonici in questo tipo di polmonite.
Amidarone
idroclorite. L'amiodarone è stato in origine usato a bassi dosaggi
terapeutici (200 mg/die) per la terapia dell'angina pectoris. Più di recente è
stato usato ad alti dosaggi (600-800 mg/die) per il trattamento delle aritmie
ventricolari refrattarie ed è stato sporadicamente riportato come causa di
tossicità polmonare, ipertiroidismo ed elevazione degli enzimi epatici. La tossicità
polmonare si manifesta, come una fibrosi che coinvolge più frequentemente i
lobi superiori. Ai Rx si presentano, infiltrati interstiziali e a chiazze negli
spazi aerei con broncogrammi aerei. In alcuni studi è stato difficile escludere
il ruolo contributorio di uno scompenso cardiaco associato. Sebbene non sia
stata riportata l’evidenza di laboratorio di un sottostante meccanismo immune,
in alcuni casi di polmonite da amiodarone sono stati somministrati ai pazienti
dei cortisonici.
Radiazioni. Il danno può
derivare da una terapia radiante applicata ai polmoni, al mediastino o alla
parete toracica. Lo sviluppo, di una malattia clinicamente significativa sembra
dipendere non solo dalla dose di radiazioni, ma anche dal volume di polmone
entro il campo irradiato. Dosi di radiazioni inferiori a 200 rad di solito non
causano polmonite. L'effetto di dosi più massicce può essere migliorato con il
frazionamento delle radiazioni. La polmonite da radiazioni e di solito
conseguenza di dosi polmonari superiori a 2000 rad. Si stima che dal 5 al 15%
dei pazienti che ricevono un'irradiazione toracica sviluppi una polmonite
clinica, e che meno del 5% muoia per conseguenze dirette del progressivo
deterioramento. In malattie come quella di Hodgkin, in cui sono somministrate
grandi dosi di radiazioni, consentendo una maggiore sopravvivenza, l'incidenza
da polmonite può raggiungere il 50%.
Anatomia patologica.
Il danno da radiazioni nei tessuti polmonari inizia nelle cellule che si
dividono più rapidamente, come i pneumociti di tipo II,
le cellule endoteliali e quelle dell'epitelio bronchiale. I cambiamenti
citologia includono edema interstiziale con infiltrati di mononucleati e
desquamazione delle cellule epiteliali alveolari e proliferazione di pneumociti
II con depositi ricchi
di fibrina e membrane ialine. Le lesioni vascolari sono comuni ed appaiono come
ingrandimento e trombosi dei capillari polmonari. È infine osservata una
deposizione interstiziale di collagene che porta alla fibrosi. Le cellule
flogistiche sono caratteristicamente assenti in questo stadio.
Fisiopatologia. Durante la
fase acuta della polmonite di solito 6-12 settimane dopo l'irradiazione un
difetto restrittivo può essere dimostrato da riduzioni di CV, CFR e CPT.
La compliance statica e la DLCO sono ridotte. Queste modificazioni
possono risolversi con un miglioramento clinico o mostrare un progressivo
deterioramento in un arco di anni. Si possono sviluppare vari gradi di
ipossiemia arteriosa senza ipercapnia.
Caratteristiche cliniche.
I sintomi iniziano di solito 6-12 settimane dopo l'irradiazione. I primi
sintomi sono di solito tosse e febbre, mentre la dispnea si sviluppa in
seguito. Dopo la fase acuta, la maggior parte dei pazienti mostra una
remissione clinica, sebbene alcuni siano interessati per altri 6-12 mesi.
Numerosi fattori possono influenzare lo sviluppo e il conseguente decorso di
una polmonite da radiazioni, tra cui una concomitante chemioterapia con citotossici, radiazioni ripetute e la sospensione
della terapia steroidea. Una ricorrenza locale, o una diffusione metastatica
della neoplasia primitiva, è difficile da differenziale da una polmonite da
raggi e può richiedere una biopsia polmonare a scopo diagnostico. Se i sintomi
ed i segni radiografici si sviluppano più di 4-6 mesi dopo la fine della
radioterapia, è più probabile una ricomparsa neoplastica. D'altro canto una
risposta favorevole del tumore primitivo alle radiazioni, seguita subito dopo
da infiltrati interstiziali, suggerisce una polmonite da radiazioni come la
diagnosi più probabile.
Radiografia del torace.
Raramente un'iperlucentezza dell'area irradiata si può osservare subito dopo la
lesione da raggi: essa può riflettere la lesione vascolare osservata
istologicamente. Più comunemente un aspetto a superficie vetrosa o una nebulosità
diffusa con strie lineari distinte (corrispondenti al campo irradiato) può
venire osservata e può aiutare a distinguere una polmonite da radiazioni da
altri processi infiltrativi come la linfangite carcinomatosa. Con l'andar del
tempo possono svilupparsi fibrosi progressiva, cicatrici locali e retrazionare
polmonare che ricordano le modificazioni osservate nella TBC cronica inattiva.
Terapia. Sebbene non provata da studi controllati,
la maggior parte degli esperti raccomanda una terapia steroidea terapia
(prednisone, 40-100 mg al giorno) durante la fase acuta della polmonite.Gli
steroidi non hanno effetto su fibrosi o cicatrici stabilizate. La terapia
anticoagulante è stata proposta a causa della trombosi microvascolare osservata
istologicamente, sebbene i benefici di questa forma di trattamento non siano
stati confermati.
Uremia.Una polmonite interstiziale diffusa può
svilupparsi nell'uremia cronica, indipendentemente dal sovraccarico idrico
dell'insufficienza renale acuta. Un'alveolite è di solito associata con gradi
significativi di fibrosi. Ai Rx vi sono infiltrati interstiziali diffusi ad
ambo le basi ed infiltrati irradiantesi dagli ili. Si possono rilevare altre
manifestazioni cliniche dello stato uremico, come prurito, pericardite e fame
d'aria dovuta all'acidosi metabolica. Con l'avvento della nefrodialisi,
raramente si osserva un'uremia cronica scompensata.
Inalazione di
polvere fibrogena Inorganica. L'inalazione
di polvere fibrogena inorganica e la causa della maggior parte delle malattie
polmonari da inalazione di polveri industriali, note complessivamente come
pneumoconiosi. Questo gruppo, comunemente assodato all'inalazione di amianto o
silice, viene preso in esame più avanti nel capitolo 78.
Fibrosi interstiziale a causa sconosciuta
Fibrosi
polmonare Idiopatica (alveolite fibrotica criptogenetica). La
fibrosi polmonare idiopatica (FPI) è una malattia infiammatoria del parenchima polmonare
caratterizzata da infiltrazione cellulare, restringimento dell'interstizio
alveolare e, talvolta, presenza di cellule mononucleate negli spazi aerei
alveolari. In casi particolarmente acuti può essere osservato un essudato
cellulare e fibrinoso con formazione di membrane ialine. Nelle forme croniche
vi sono meno manifestazioni essudative e una maggiore fibrosi. La presentazione
clinica varia da una polmonite interstiziale acuta fulminante (s.di Hamman-Rich
entità morbosa rarissima) a una fibrosi progressiva che porta a un polmone in
fase terminale. Sebbene la FPI sia stata inizialmente una diagnosi di
esclusione, attualmente essa è considerata, in genere, una specifica entità
patologica ad eziologia sconosciuta.
{kind=link}
Classificazione. La
FPI può essere classificata in base ai suoi caratteri istologici come:
polmonite interstiziale, classica o comune" (PIC) polmonite
interstiziale desquamativa (PID), in
cui vi è una proliferazione di macrofagi e desquamazione dei pnumociti
granulari negli spazi alveolari e nelle vie aeree terminali polmonite
interstiziale linfoide (PIL), in cui l'interstizio e gli spazi alveolari
sono infiltrati da cellule linfoidi polmonite interstiziale a cellule giganti (PIG),
con infiltrato cellulare misto comprendente cellule giganti fagocitiche
irregolari. In pratica, tuttavia, possono coesistere più tipi istologici o vi
può essere trasformazione di un tipo nell'altro. Di conseguenza, la risposta
agli steroidi e la prognosi relativamente migliore osservate nella variante PID
sono credute da alcuni una prova che la PID stessa rappresenti rispetto alle altre varianti uno stadio
precoce di FPI.
Eziologia a
patogenenesi. Una vasta gamma di reperti associati di laboratorio ha
suggerito un'eziologia da ipersensibilità o autoimmune. Nella FPI sono state
dimostrate iperglobulinemia, test di Coombs positivo, autoanticorpi,
criglobuline, fattore reumatoide ed eosinofilia. Il processo fibrotico è
preceduto da cambiamenti infiammatori cronici, caratterizzati da infiltrazione parenchimale di
linfociti, macrofagi, neutrofili ed eosinofili. Al microscopio si nota l'aumento di deposito di collagene nei setti
polmonari, sebbene i dati quantitativi sulla quota di collagene e la sua
frequenza di sintesi siano entro i limiti normali. Ulteriori studi hanno
rivelato variazioni qualitative del collagene del tessuto connettivo in termini
di proporzione tra collagene
di tipo I e III. Il collagene di tipo I è aumentato in rapporto al tipo III e di conseguenza le pareti alveolari hanno una
minore compilante. Il collagene di tipo I impregna di preferenza le colorazioni usate per il tessuto
connettivo, portando all'impressione di un aumento complessivo del collagene
polmonare alla microscopia ottica. La microscopia elettronica mostra che
le fibre collagene diventano logore e contorte, variazione che si associa alla
presenza di collagene entro il liquido broncoalveolare di alcuni pazienti con
FPI. I neutrofili hanno forse un ruolo importante nel processo flogistico ed
insieme ai macrofagi (che possono forse secernere un fattore chemiotattico per
i neutrofili) essi iniziano e sostengono il danno polmonare e la fibrosi. I
linfociti possono contribuire alla secrezione di un fattore inibente la
migrazione dei macrofagi, che immobilizzati secernono in seguito uno specifico
fattore chemiotattico per i neutrofili.
Fisiopatologia.
Insieme ad altre malattie polmonari interstiziali fibrotiche, i disturbi della
funzione polmonare sono caratterizzati da una riduzione di CV, CFR e
CPT. Una parte significativa (dal 50 al 60%) mostra segni evidenti di
malattia delle piccole vie respiratorie dimostrabile dalla dipendenza della
compliance dalla frequenza respiratoria e dalla diminuzione dei flussi
espiratori forzati a bassi volumi polmonari. Questi ultimi cambiamenti danno
luogo ad un cattivo rapporto ventilazione/perfusione, che può spiegare la
maggior parte dell'ipossiemia arteriosa nei pazienti. Si osservano anche
riduzioni associate della DLCO, ed esse possono essere utilizzate per
controllare il decorso clinico della malattia. Valori di DLCO al di
sotto del 45% del predetto valore sono comunemente associati ad ipertensione
polmonare e a cuore polmonare.
Segni clinici.
Molti pazienti presentano dispnea durante sforzo od anche a riposo. Si sviluppa
spesso una tosse secca, non produttiva, sebbene nelle forme acute e subacute
possa essere presente espettorato. Una crepitazione bilaterale è udibile alle
basi polmonari dei pazienti, di cui il 40% circa presenta un ippocratismo
digitale. In genere viene riportata una leggera prevalenza nel sesso maschile,
con età di comparsa dai 12 ai 63 anni. Titoli positivi per il
fattore reumatoide sono stati descritti dal 20% al 60% dei casi e l'ANF è presente nel 30-40% dei casi. L'esame del
contenuto cellulare del liquido ottenuto con un lavaggio broncoalveolare svela
un 10-20% di neutrofili, in contrasto con la predominanza linfocitaria
osservata nella sarcoidosi o nella polmonite da ipersensibilità. La biopsia
transbronchiale può essere utile alla diagnosi, che è però limitata a causa del
piccolo numero di biopsie e degli errori di campionamento inerenti a questa
tecnica. Per stabilire la diagnosi può perciò essere necessaria una biopsia
polmonare a cielo aperto.(Al momento della biopsia
polmonare a cielo aperto occorre richiedere al chirurgo più campioni prelevati
dalla parte più colpita, è dalla parte meno colpita del polmone. In questo modo
si può meglio valutare lo spettro degli aspetti istologici presenti).Un deterioramento rapido e progressivo che porta a morte
precoce si può osservare in una piccola percentuale dei pazienti, secondo il
decorso descritto nel primo studio di Hamman e Rich. Nonostante molti casi
mostrino una maggiore sopravvivenza, il 50% dei pazienti muore entro 6/12
anni dalle prime manifestazioni.
Radiografia dal
torace. In relazione
allo stadio ed alla progressione della fibrosi sono stati osservati quadri
variabili. Nelle fasi precoci della malattia è spesso osservabile un fine
quadro reticolare alle basi. Queste alterazioni possono progredire
verticalmente lungo i campi polmonari, divenendo reticolonodulari, per
svilupparsi alla fine in una cicatrizzazione fibrosa con aspetto ad alveare.
Con la riduzione dei volumi polmonari il diaframma risale progressivamente. Un
interessamento pleurico e linfonodale e raro è quando è presente suggerisce una
patologia diversa e coesistente.
Terapia.
Sembra che i corticosteroidi
rallentino il progresso della malattia, sebbene la prognosi rimanga infausta
gli effetti della terapia si evidenziano meglio durante lo stadio di alveolite
attiva, poiché i corticosteroidi non hanno alcun effetto sulla fibrosi già
instaurata. Inizialmente la variante PID risponde
meglio della PIC o di altre varianti. I farmaci
immunosoppressori da soli purtroppo sono inefficaci e non mostrano alcun
effetto benefico quando vengono somministrati assieme ai corticosteroidi. L'uso della penicillamina, un farmaco che
inibisce il cross-linking (unione incrociata) del collagene, attende una
valutazione più precisa.
(PS. Come si
può leggere in questo capitolo di malattie interstiziali n’esistono tantissime
e la maggior parte di queste la sua natura può essere conosciuta, e accertata
con tante indagini diagnostiche complesse prima di iniziare delle terapie.
Nella storia che vide protagonista Adriana tutto questo non e stato fatto, la
stessa biopsia polmonare non e stata fatta in modo corretto, cioè prelevando
più pezzettini di tessuto polmonare per avere una visione molto più chiara
della situazione, gli accertamenti diagnostici fatti sono molto superficiali,
non e stato fatto neanche un banalissimo esame dell’ispettorato che e molto
importante. Non sto qui a scrivere quello che non si e fatto in quanto mi pare
che questo lo gia scritto, dico solo che non e stato fatto nulla per Adriana.
Dal vetrino della biopsia polmonare in vivo che io ho fatto esaminare da vari
patologi quello che e stato visto e una forma di polmonite, con alcune cellule
squamose, e in ogni modo mi e stato affermato che non si può fare una diagnosi
basandosi solo su questo vetrino, occorrono altre indagini per avere una
certezza della malattia. ( ora io mi domando sé! da questo vetrino non si può
fare diagnosi di certezza della patologia; come hanno fatto i medici al suo
tempo a fare diagnosi di fibrosi polmonare tipo (UIP?). Sé si esaminano i
vetrini della autopsia probabilmente può esserci una forma fibrotica, ma questa
e stata portata dalle condizioni di come e stata tenuta, e trattata, dalle
infezioni batteriche, e altro che si e presa cioè, le cosi dette infezioni
opportunistiche del paziente immunodepresso a causa di farmaci ho altro, è dai
farmaci che gli hanno somministrato, cioè i farmaci citotossici, infatti, come
si può leggera dalla letteratura medica e noto che i farmaci possono causare
una malattia interstiziale, polmoniti, è fibrosi polmonari; e specialmente i
farmaci citotossici, ho gli immunosoppressori. Dall’istologico dell’autopsia si
legge che c’è infiammazione cronica nell’interstizio e una marcata
proliferazione fibroplastica. Questo può essere coerente con la letteratura
medica, come mai però tutto questo, nell’istologico della biopsia in vivo non e
coerente con la letteratura medica? Queste malattie hanno delle caratteristiche
ben precise, per poi formarsi, per come sono descritte dalla letteratura medica
in campo internazionale, e ci sono determinate cellule, coinvolte nelle
fibrosi, Infiammazione, fibroblasti in gran numero, ecc. e lo stesso collagene
mi pare che sia di un determinato tipo per formare il tessuto fibrotico;
giusto? (esempio. prima che si metta a piovere nell’atmosfera terrestre si
formano i vapori acquei, e poi può piovere, sé manca uno solo di tutti gli
elementi che occorrono per formare l’acqua; non può esserci pioggia. Mi sembra
che questo concetto sia abbastanza chiaro, oppure no?). Io à queste e a tante
altre domande voglio una risposta!. È la verità può essere accertata con tutte
le indagini mediche legali che sono da farsi.